
More Than Just a Hangover
Alcohol consumption is responsible for a significant global disease burden and caused over 2.4 million deaths in 2019. It has been estimated that alcohol-associated chronic liver diseases accounted for 11.2 million disability-adjusted life years (DALYs) in the same year.[1,2] The consumption of alcohol appears to be increasing over time according to Global Burden of Disease data and has been accompanied by a concurrent reduction in alcohol abstinence. Both of these trends are projected to continue to at least 2030. Heavy episodic alcohol consumption, defined as at least 60 g alcohol in one or more episodes over 30 days, is also increasing with 18.5% of adults consuming alcohol in this manner in 1990 and 20% in 2016. This has been projected to increase to 23% in 2030.[3]
The exact definition of ‘binge drinking’ is unclear. The National Institute on Alcohol Abuse and Alcoholism (NIAAA) defines binge drinking as alcohol consumption that increases blood alcohol concentration to 0.08 g/dL or greater, which equates to four or more drinks for females over 2 hours, or five or more drinks for males.[4] This definition is mentioned in guidelines by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver.[5,6] However, this does not take into account the total amount consumed in a binge drinking episode. Indeed, the precise role of binge drinking in the progression to advanced liver disease has not been definitely established. A Danish study suggested that daily drinking posed a greater risk of cirrhosis specifically in men,[7] while a study from Finland concluded that binge drinking was associated with a higher risk.[8] Sex disparity may also influence the progression to cirrhosis, with a recent analysis of the UK Million Women Study suggesting that daily drinking and alcohol consumption without meals were associated with the highest risk in middle aged women.[9] A consistent issue with the literature, which largely consists of epidemiological studies, is that alcohol consumption is usually self-reported and hence is prone to bias.
Conversely, the association of histological fibrosis with progression of alcohol-associated liver disease is well-described.[10] Pericellular elastosis appears to be a hallmark of more severe, established disease.[11] Recently, collagen proportionate area has been demonstrated to predict clinical outcomes,[12] as has a semi-quantitative histological scoring system.[13] However, the association between fibrogenesis and the pattern of alcohol consumption has not been explored in depth. This is one of several aspects in the disease course of alcohol-associated liver disease where there is a need for the development of prognostic biomarkers (Figure 1).
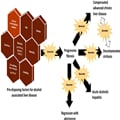
Figure 1.
The disease course of alcohol associated liver disease highlighting the potential areas where prognostic biomarkers may be useful. The study by Torp and colleagues14 examines the predisposing factor of alcohol consumption pattern and progressive fibrosis. Abbreviations: NAFLD, non-alcoholic fatty liver disease
In this issue of Liver International, Torp and colleagues[14] investigate the impacts of binge drinking on serum markers of hepatic extracellular matrix (ECM) deposition and degradation in a proof-of-concept study using a unique methodology. Patients (N = 29) with alcohol-associated liver disease (ALD) who were actively drinking and patients with histologically characterised non-alcoholic fatty liver disease (NAFLD) were stratified by the presence of significant fibrosis (≥F2) and compared to 10 healthy controls. To simulate binge drinking, 40% ethanol in 9mg/ml NaCl solution was administered via nasogastric tube at a dose of 2.5mL/kg body weight over 30 minutes. Those with BMI >25 kg/m2 were given a dose adjustment of 0.5 mL/kg to avoid severe intoxication. Blood was drawn during the intervention from the hepatic vein and the right external jugular vein and at multiple time points up to 3 hours. Further peripheral blood sampling occurred at 24 hours. Markers of collagen formation (PRO-C3, PRO-C4 and PRO-C8) and degradation (C3M and C4M) were then measured using competitive ELISA assays. All participants abstained from alcohol for at least the 48 hours prior to the intervention.
The mean peak blood alcohol concentration was 34 mmol/L (0.16 g/dL) after 1 hour and 21 mmol/L (0.10 g/dL) after 3 hours, satisfying the NIAAA definition of binge drinking. Interestingly, in all participants PRO-C3 increased by 10% from baseline at 24 hours. Indeed, the authors termed the observed PRO-C3 response a ‘burst’, given that the basal turnover of type III collagen is 0.2%-0.6% per day in comparison. In patients with significant fibrosis, systemic PRO-C3 levels decreased in the first 3 hours, while there were no significant changes in healthy controls and those with F0-F1 fibrosis. The authors suggest this may be because of fibrosis remodelling in patients with fibrosis and that degradation of existing type III collagen precedes deposition. However, comparative increases in C3M to support this theory were not observed. Indeed, those with baseline significant fibrosis had higher hepatic vein type III collagen formation vs degradation (PRO-C3/C3M ratio) compared to those with F0-F1 fibrosis (P = .006) and healthy controls (P = .003). However, this change was not seen in systemic blood samples. C3M and C4M at 24 hours did not show any change from baseline. Changes between sites of blood sampling as well as other markers of collagen formation and degradation were less informative from this study.
This interesting study confirms that a standardised dose of alcohol that accurately simulates an acute alcohol binge can trigger a fibrogenic response at 24 hours in both healthy individuals and those with chronic liver disease, illustrated by an increase in PRO-C3 levels. PRO-C3 is the N-terminal pro-peptide of type III collagen and has been correlated with histological fibrosis independently, but also as part of combined models in viral hepatitis, ALD and NAFLD.[15–17] Markers of fibrosis degradation did not increase concomitantly. There are some significant limitations in the interpretation of the results of the study, particularly that hepatic vein blood concentrations were not available at 24 hours, nor were hepatic vein blood alcohol concentrations presented. Furthermore, beyond the associations mentioned, there was no attempt to characterise the pathobiology of the PRO-C3 response. Specifically, the investigation of the role of inflammatory cytokines and chemokines, endothelial activation, bacterial translocation as well as reactive oxygen species may characterise this response in detail. Moreover, the kinetics of PRO-C3 and other ECM markers beyond 24 hours was not demonstrated, which is particularly pertinent as blood alcohol concentrations become undetectable.
The implication of this study is that it establishes the relationship between binge drinking and the potential for fibrogenesis and progression to advanced liver disease. Strikingly, the PRO-C3 response was also observed in healthy controls. This suggests that PRO-C3 may have potential as a biomarker in terms of response to abstinence and regression of significant or advanced fibrosis (Figure 1). Regardless, the longer term kinetics of PRO-C3 and other ECM markers require further study, as do the underlying mechanisms of collagen deposition in acute alcohol consumption. This may eventually lead to further understanding of what amount of alcohol, if any, may be considered ‘safe’ in people with co-existing liver diseases such as NAFLD. Ultimately, the authors should be commended on shedding new light on this important topic and that the consequences of binge drinking may be more than just a hangover.